


1細胞マルチオームの実験と得られるデータ形式
目次
実験手順の概要
1. scATAC-seq用の前処理
-
細胞膜を破砕し核を抽出。
-
Tn5トランスポザーゼを加えてクロマチンの開いた領域を切断し、両端にアダプター配列を付加した断片を核内に作成。
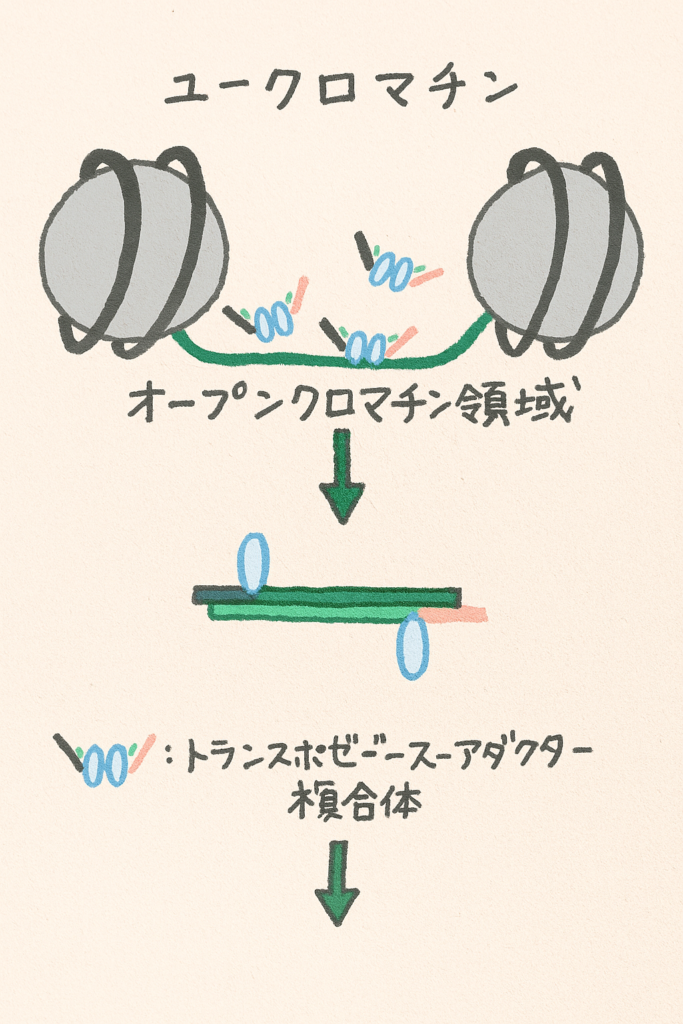
2. ライブラリ調製
-
アダプター配列付きオープンクロマチンの断片を含む核1つに対して1つのGEMを作成。このステップで1細胞毎に分ける。
-
核内のmRNAをGEMに含まれるオリゴ(dT)プライマーで捕捉し、逆転写反応でcDNAを合成。このステップで細胞識別用のバーコードを付加する。
-
ATACでの前処理で得られたアダプター配列付き断片に対してもPCRを利用して細胞識別用のバーコードを付加。
-
上記のcDNAとATAC断片由来に付加するバーコードは同じ核由来であれば同一。これによりトランスクリプトームとオープンクロマチンの情報が同じ細胞由来として結びつけられる。
-
PCRを通してNGSによるシーケンスに必要なP5、P7などの配列を付加したライブラリを調製する。
3. シーケンスとデータ解析
-
mRNA由来のライブラリとATAC用のライブラリそれぞれについてNGSによるシーケンスを実施。
-
CellRanger ARCによってNGS由来のfastqデータがMatrix(行が細胞で列が遺伝子またはゲノム領域であるイメージ)に変換される。
-
研究者が実施するバイオインフォマティクスの処理はMatrixデータを起点に実施していくのが通常のワークフロー。

実験手順の詳細
Step 1: 核の単離(Nuclei Isolation)
原理
本キットでは細胞膜を選択的に破壊し、核を損傷なく回収することが重要です。RNAとクロマチン情報を同時に取得するため、RNAの分解を抑えつつ、クロマチン構造を保持する条件で核を単離します。
手順
-
細胞をPBSで洗浄し、遠心(300–500 g, 5分)。
-
**氷冷の核抽出バッファ(Nuclei Isolation Buffer)**に再懸濁し、数分間インキュベーション。
-
Triton X-100 や NP-40を含む洗浄バッファで細胞膜を溶解。
-
70 µmフィルターで濾過し、核を精製。
-
顕微鏡で核の形態・破損率を確認。
-
核数を計測し、最終濃度は 300–1200 nuclei/µL に調整。
Step 2: Tn5トランスポジション(Chromatin Accessibility Tagging)
原理
Tn5トランスポザーゼが開いたクロマチン領域(オープンクロマチン)に結合し、DNAを切断しながらアダプター配列を挿入します(tagmentation)。
手順
-
単離した核にTn5 transposase mixを添加。
-
37℃で5分間インキュベート(キット推奨条件)。
-
反応を氷上で停止し、次のGEMステップへ進行。
Step 3: GEM生成(Gel Bead-in Emulsion)
原理
GEM(Gel Beads-in-Emulsion)は1細胞(または核)あたり1つのゲルビーズと反応液を閉じ込めた油中水滴で構成されます。ビーズにはRNA用およびATAC用のバーコード化オリゴが付与されています。
-
RNA: 5' - P7 - 10X Barcode - UMI - poly(dT) - 3'
-
ATAC: 5' - P5 - Tn5 adaptor - 10X Barcode - 3'
手順
-
核+トランスポジション済DNAを10Xランチャーにロード。
-
Chromium Next GEMチップに試薬類と一緒にセット。
-
クロマチックエンカプセル化により、GEMが自動的に形成。
-
各GEM内では:
-
RNAがバーコードオリゴ(dT)に捕捉
-
同時に、ATAC断片とバーコードが結合
-
Step 4: 逆転写反応とバーコード付与(Reverse Transcription and Barcoding)
原理
GEM内でmRNAが逆転写され、cDNAが10X固有のバーコード(+UMI)とともに合成されます。これにより、後工程で細胞由来の識別が可能になります。
手順
-
GEMを42℃で逆転写反応(RT)インキュベート(~2時間)。
-
エマルジョンをブレークし、バルクcDNAを精製(silica columnなど使用)。
-
cDNAはATACライブラリと分離されて保持。
Step 5: PCR増幅(Library Amplification)
原理
RNA由来cDNAとATAC断片は、それぞれバーコード情報を保持した状態でPCR増幅されます。
手順
RNA部位:
-
SPRI beadsで精製後、cDNAを一次PCRにより増幅(14–16サイクル程度)。
-
二次反応でP5/P7アダプターを付加して完成。
ATAC部位:
-
Tagmentation済DNAをSPRIで精製。
-
PCRで増幅し、同様にP5/P7アダプターを付加。
Step 6: ライブラリ精製・QC(Quality Control)
原理
サイズ選別や不純物除去を行うことで、**正しいサイズレンジ(RNA: ~400 bp、ATAC: ~300–600 bp)**のライブラリを取得します。
手順
-
AMPure XP beadsでライブラリサイズ選別。
-
Agilent BioanalyzerやTapeStationでライブラリのサイズ・濃度評価。
-
qPCRでライブラリ定量(KAPA kit推奨)。
Step 7: シーケンス(Illumina platform)
原理
RNAおよびATACそれぞれのライブラリは独立にシーケンスされ、同一バーコードを使って解析上で統合されます。
推奨条件
RNA-seqライブラリ
-
Paired-end 28 bp (R1) + 90 bp (R2)
-
R1: cell barcode + UMI
-
R2: transcript
ATAC-seqライブラリ
- Paired-end 50 bp (R1/R2)
Step 8: データ解析(Cell Ranger ARC)
原理
Cell Ranger ARCを用いて、RNAとATACデータをそれぞれマッピング・フィルタリング・細胞の同定を行います。さらに、同一細胞のRNAとクロマチンの統合解析が可能です。
主な出力
-
filtered_feature_bc_matrix (トランスクリプトーム)
-
barcodes.tsv.gz (細胞毎に付与されたバーコード一覧)
-
features.tsv.gz (遺伝子名一覧)
-
matrix.mtx.gz (リードカウント数の行列データ(実際は「行数、列数、カウント数」からなる圧縮された形式))
-
-
filtered_feature_bc_matrix.h5
-
atac_fragments.tsv.gz
-
peak_annotation.tsv
-
gene_activity_matrix
次の記事
次の記事ではscnapyを用いたscRNA-Seqデータの前処理について説明します。