


【ADHD血液診断】交差検証等を実施した最終モデル
目次
- 0. はじめに
- 1. 交差検証・グリッドサーチを実施しながらモデル構築を繰り返す
- 2. 精度
- 3. ROCカーブ
- 5. AUC
- 6. feature_importance_
- 7. おまけ
- 8. まとめ
0. はじめに
本記事は、以下の記事の続きになります。
https://www.bioinforest.com/adhd1-5
1. 交差検証・グリッドサーチを実施しながらモデル構築を繰り返す
Text
import timeimport pickle
import pandas as pdimport numpy as np
import matplotlib.pyplot as pltimport seaborn as sns
from scipy.stats import pearsonrfrom sklearn.decomposition import PCAfrom sklearn.manifold import TSNEimport umapfrom scipy.sparse.csgraph import connected_components
import scipy.stats as stfrom scipy.stats import mannwhitneyu
from sklearn.model_selection import train_test_splitfrom sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import accuracy_scorefrom sklearn.metrics import roc_curvefrom sklearn.metrics import roc_auc_score
from sklearn.model_selection import cross_val_scorefrom sklearn.model_selection import StratifiedKFoldfrom sklearn.model_selection import GridSearchCV
import warningswarnings.filterwarnings('ignore')
# パラメータを dict 型で指定search_params = { 'n_estimators':[i for i in range(10, 210, 20)], 'criterion': ['entropy', 'gini'], 'max_features':[i for i in range(1, 10, 2)], 'max_depth':[i for i in range(1, 10, 2)],}
def random_forest(X, y, search_params): t0 = time.time()
X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, stratify=y)
model = RandomForestClassifier() stratifiedkfold = StratifiedKFold(n_splits=5) grid_search = GridSearchCV(model, search_params, cv=stratifiedkfold) grid_search.fit(X_train, y_train)
best_model = grid_search.best_estimator_ score = grid_search.score(X_test, y_test) prob = list(grid_search.predict_proba(X_test)[:,1]) fi = list(grid_search.best_estimator_.feature_importances_) best_param = grid_search.best_params_
t1 = time.time() elapsed_time = t1 - t0
return best_model, score, prob, list(y_test), fi, best_param, elapsed_time
models = []scores = []probs = []y_tests = []fis = []best_params = []
for i in range(200): res = random_forest(X, y, search_params)
models.append(res[0]) scores.append(res[1]) probs.extend(res[2]) y_tests.extend(res[3]) fis.append(res[4]) best_params.append(res[5])
print(f"Elapsed time for iteration {i+1} was {res[6]:.3f} sec. Accuracy score was {res[1]:.5f}.")Text
Elapsed time for iteration 1 was 199.041 sec. Accuracy score was 0.68750.Elapsed time for iteration 2 was 199.286 sec. Accuracy score was 0.68750.Elapsed time for iteration 3 was 199.067 sec. Accuracy score was 0.75000.Elapsed time for iteration 4 was 199.194 sec. Accuracy score was 0.87500.Elapsed time for iteration 5 was 199.040 sec. Accuracy score was 0.75000....Elapsed time for iteration 199 was 199.306 sec. Accuracy score was 0.93750.Elapsed time for iteration 200 was 198.992 sec. Accuracy score was 0.87500.2. 精度
Text
plt.hist(scores, bins=50)plt.show()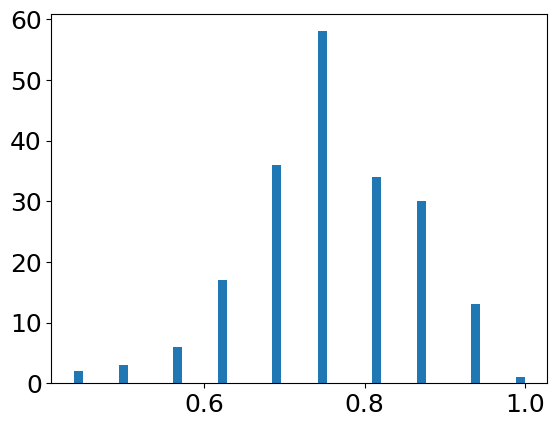
Text
mean = np.mean(scores)std = np.std(scores)print(f"accuracy: {mean:.3f} ± {std:.3f}")Text
accuracy: 0.758 ± 0.1033. ROCカーブ
Text
fpr_all, tpr_all, thresholds_all = roc_curve(y_tests, probs, drop_intermediate=False)
plt.plot(fpr_all, tpr_all, marker='o')plt.xlabel('FPR: False positive rate')plt.ylabel('TPR: True positive rate')plt.grid()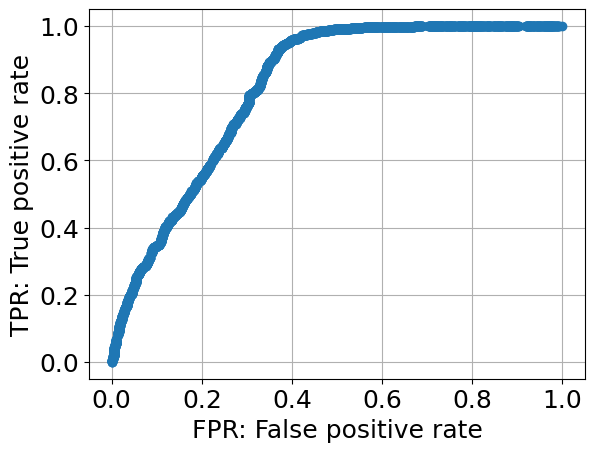
5. AUC
Text
roc_auc_score(y_tests, probs)Text
0.81624121093756. feature_importance_
Text
df_fis = pd.DataFrame(fis, columns=df_ttest.columns.tolist()[:-1])
fis_gene_names = df_fis.columns.tolist()fis_means = df_fis.apply(lambda x: np.mean(x)).tolist()fis_stds = df_fis.apply(lambda x: np.std(x)).tolist()
fis_means_sorted_idxes = np.argsort(fis_means)[::-1][:50]
x = [fis_gene_names[i] for i in fis_means_sorted_idxes]y = [fis_means[i] * 100 for i in fis_means_sorted_idxes]std = [fis_stds[i] for i in fis_means_sorted_idxes]
plt.rcParams["figure.figsize"] = (20,3)plt.bar(x, y, yerr=std)plt.xticks(x, fontsize=10)plt.xticks(rotation=90)plt.xlabel('Gene Name')plt.ylabel('Score')plt.show()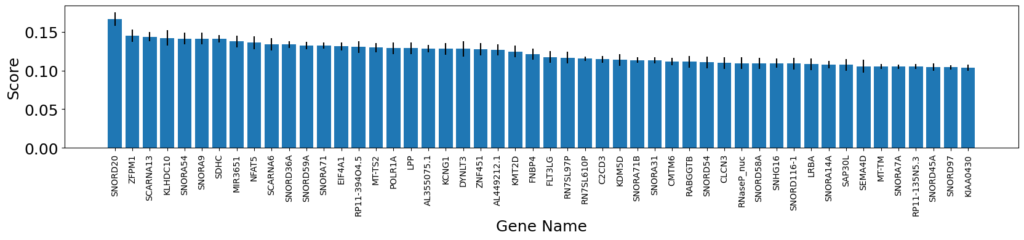
7. おまけ
確率計算を下記のように工夫すると、ほぼ100%当たるモデルができます。
Text
def model_final(models, scores, transcriptome): probs = []
for i in range(len(models)): ps = models[i].predict_proba(np.array(transcriptome).reshape(1, -1)) v1 = scores[i] * ps[0, 1] v2 = (1 - scores[i]) * ps[0, 0] probs.append(v1 + v2)
probs_mean = np.mean(probs) probs_std = np.std(probs)
return probs, probs_mean, probs_std
res = model_final(models, scores, X_test.iloc[0,:].tolist())
probs = []
for i in range(len(df_ttest)): res = model_final(models, scores, df_ttest.iloc[i,:-1].tolist()) probs.append(res[1]) print(f"score = {res[1]:.3f} ± {res[2]:.3f}, label = {df_ttest.iloc[i,-1]}")
fpr_all, tpr_all, thresholds_all = roc_curve(df_ttest.iloc[:,-1], probs, drop_intermediate=False)plt.rcParams["figure.figsize"] = (7,7)plt.plot(fpr_all, tpr_all, marker='o')plt.xlabel('FPR: False positive rate')plt.ylabel('TPR: True positive rate')plt.grid()
roc_auc_score(df_ttest.iloc[:,-1], probs)8. まとめ
本記事で「血液トランスクリームデータからのADHD予測」は終了となります。
AUCが約0.8で予測可能なモデルは十分に有用です。
本モデルで完全にADHD診断をすることは難しいですが、現在のDMS-5に基づいた医師による診断と、血液検査と本モデルによる診断を組み合わせることで、誤診回避を改善できそうです。
以上になります。