

【備忘録】PythonのBokehでBandのグラフを描く【可視化】
目次
Bokehのインストール
Bokehを用いるので、下記等でBokehをインストールしておきます。
Text
pip install bokehライブラリ
Bokehのうち、以下のライブラリをimportします。
Text
from bokeh.models import Band, ColumnDataSourcefrom bokeh.plotting import figure, showfrom bokeh.io import output_notebook, showfrom bokeh.models import ColumnDataSource, HoverTool, LinearColorMapperfrom bokeh.plotting import figurefrom bokeh.models import SaveTool擬似データの作成
グラフの描画には、1つのx座標に対して1つのy座標の値およびその座標におけるy軸方向の標準偏差(bandの幅に相当)のデータが必要です。
そこで、以下のような関数を用意します。この関数はy座標の母平均(mu)のセットを与えるとx座標とy座標と標準偏差のセットを出力します。
Text
def generate_dataset(mu): # 正規分布の乱数生成 rng = np.random.default_rng()
# x軸の値 x = list(range(100))
# mu: 各点における母平均
# 各点における母標準偏差 sigma = [abs(rng.normal(1, 1)) for i in range(100)]
# 母平均と母標準偏差を用いて各点における1000個のデータセットを生成 data = [rng.normal(m, s, 1000) for m, s in zip(mu, sigma)]
#1000個のデータセットから平均と標準偏差を求める mean = [np.mean(d) for d in data] std = [np.std(d) for d in data]
# プロット用に標準偏差を用いてバンドの上限と下限を計算 upper = [mean[i] + std[i] / 5 for i in range(len(mean))] lower = [mean[i] - std[i] / 5 for i in range(len(std))]
# Return return x, mean, upper, loweこの関数は例えば以下のように用います。
Text
mu = [rng.normal(0.03 * i, 0.025) for i in range(100)]x, m, upper, lower = generate_dataset(mu)描画
上述の関数で擬似データを作成しながら、以下のようにしてグラフを描画します。
Text
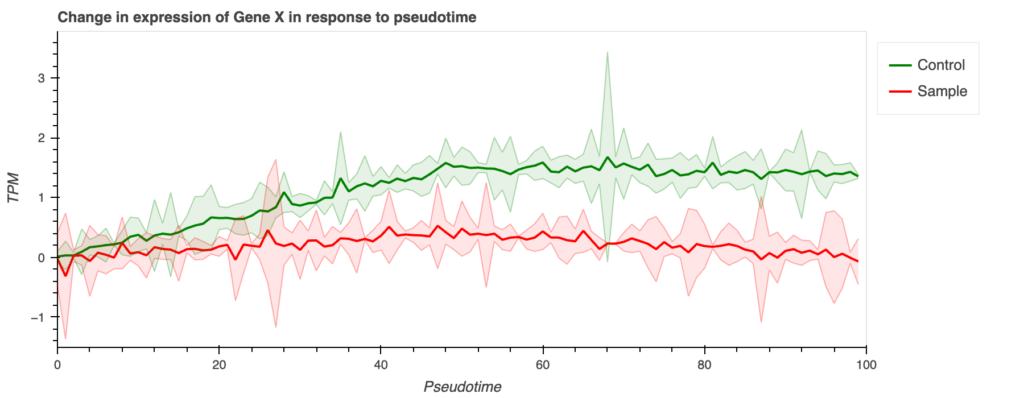
# bokeh graphp = figure(width=900, height=350, tools=["save"])p.title.text = "Change in expression of Gene X in response to pseudotime"p.xgrid.grid_line_alpha = 0p.ygrid.grid_line_alpha = 0
# axis labelp.xaxis.axis_label = 'Pseudotime'p.yaxis.axis_label = 'TPM'
# series 1mu = [rng.normal(0.03 * i, 0.025) for i in range(50)] + [rng.normal(1.5 - 0.002 * i, 0.025) for i in range(50)]x, m, upper, lower = generate_dataset(mu)source = ColumnDataSource(data=dict(x=x, m=m, lower=lower, upper=upper))p.line("x", "m", color="green", line_width=2, source=source, legend_label="Control")band = Band(base="x", lower="lower", upper="upper", source=source, fill_alpha=0.1, fill_color="green", line_color="green")p.add_layout(band)
# series 1 max/minmax_1 = max(upper)min_1 = min(lower)
# series 2mu = [rng.normal(0.008 * i, 0.025) for i in range(50)] + [rng.normal(0.4 - 0.008 * i, 0.025) for i in range(50)]x, m, upper, lower = generate_dataset(mu)source = ColumnDataSource(data=dict(x=x, m=m, lower=lower, upper=upper))p.line("x", "m", color="red", line_width=2, source=source, legend_label="Sample")band = Band(base="x", lower="lower", upper="upper", source=source, fill_alpha=0.1, fill_color="red", line_color="red")p.add_layout(band)
# series 2 max/minmax_2 = max(upper)min_2 = min(lower)
# x/y axis rangey_max = max(max_1, max_2)y_min = min(min_1, min_2)p.x_range=Range1d(0, 100)p.y_range=Range1d(y_min - 0.1*abs(y_min), y_max + 0.1*abs(y_max))
# legend positionp.add_layout(p.legend[0], "right")
output_notebook()show(p)得られるグラフの例
前述のコードで得られるグラフの例は以下です。
サムネと同じグラフを描画したい場合は、こちらの記事を参照して下さい。
サムネのグラフはsingle cell RNA-Seqのデータに対してSTREAMというツールを用いてUMAPによって次元削減した後にTrajectoryを描画し、Trajectoryを横軸にした場合の特定の遺伝子の発現量の変化を描画したものであり、描画には少々手間がかかります。